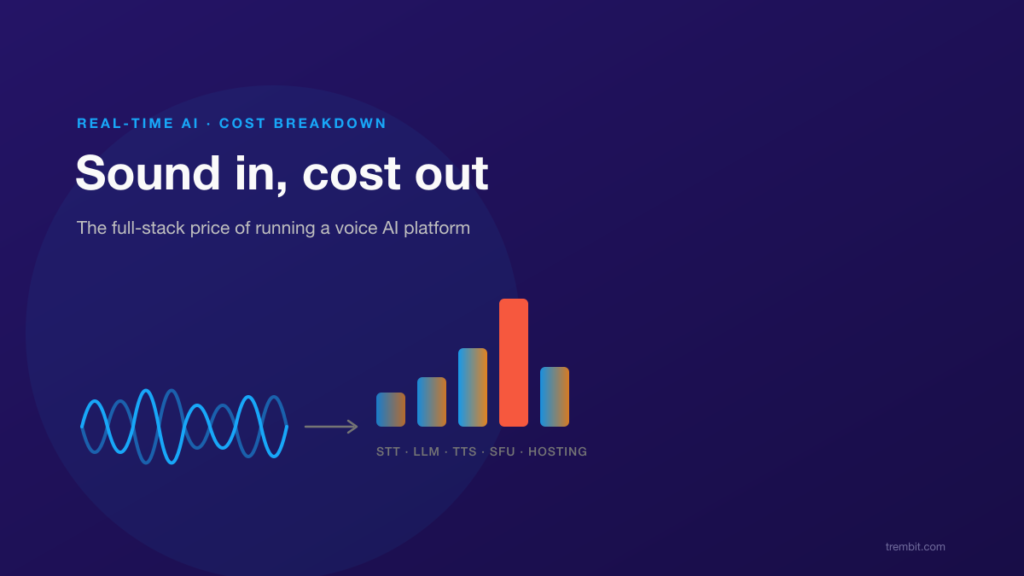
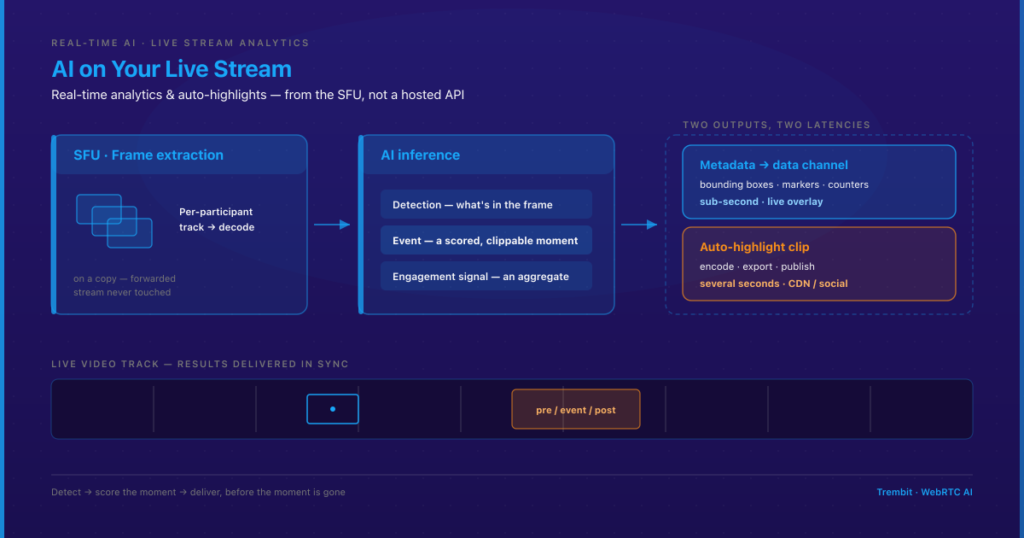
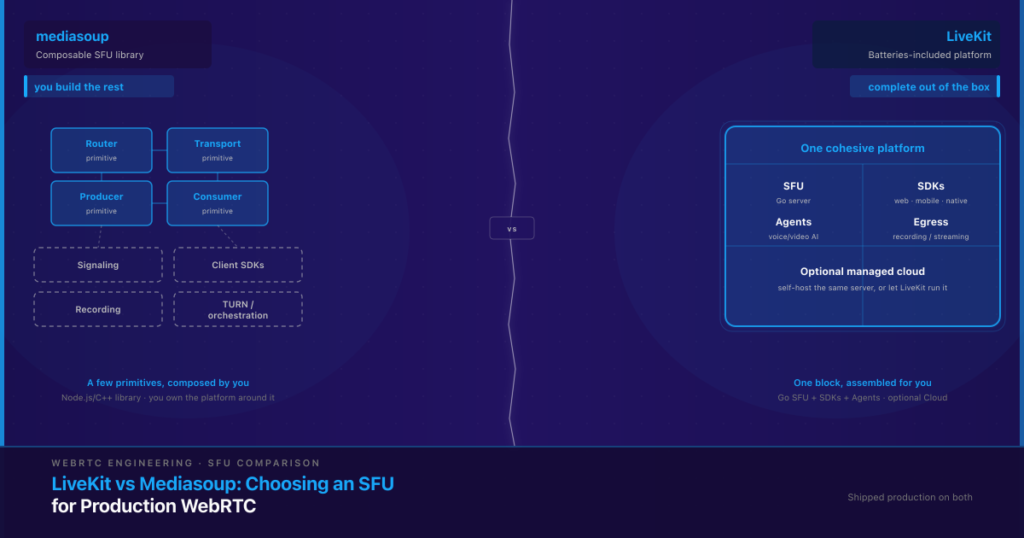
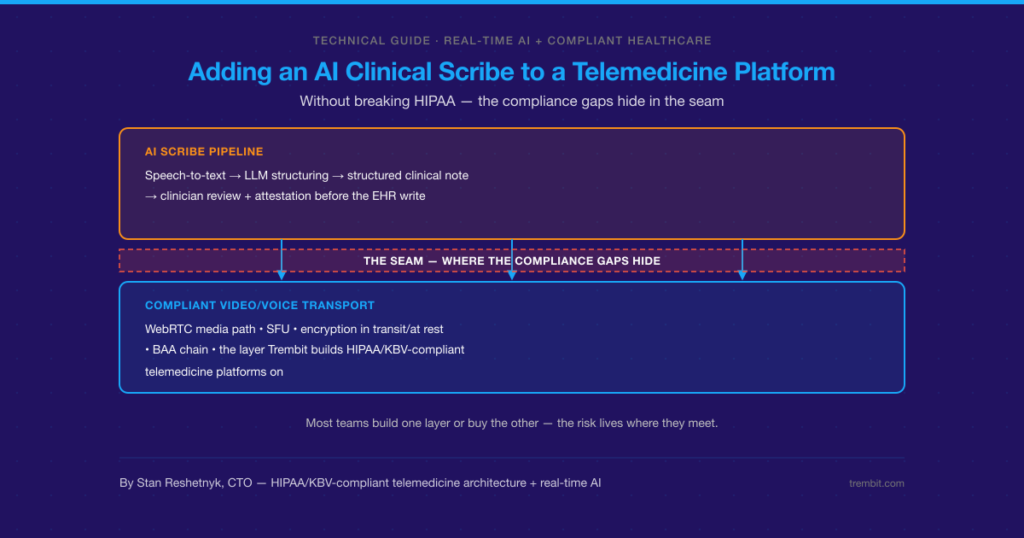
Deploying clinical AI safely means satisfying at least three overlapping obligations at once — and the cost of getting it wrong isn’t a late feature. It’s a blocked NHS or HIPAA review, a patient-safety incident, or a compliance retrofit that blows up the timeline and budget you already committed to. If the system touches NHS or UK health IT, the UK’s DCB0129 and DCB0160 clinical risk management standards apply. If it touches US patient data, HIPAA’s Security Rule safeguards and Business Associate Agreement (BAA) requirements apply. And an emerging layer of AI-specific governance — the EU AI Act and the FDA’s Software as a Medical Device framework — increasingly sits on top of both. None of these is satisfied by the model itself. They’re satisfied by the system built around it: data governance, human-in-the-loop checkpoints, audit trails, and a documented risk process.
Trembit builds that system. We’re a WebRTC and real-time video/voice engineering team that has shipped HIPAA-, GDPR-, and KBV-compliant healthcare platforms — the audit-grade infrastructure clinical AI actually runs on, whether that’s an ambient-documentation assistant riding on a telehealth call or a risk-scoring model writing into an EHR. We’re the engineering partner that helps teams meet these standards — not a Clinical Safety Officer, not a regulatory authority, and not a diagnostic-model vendor.
Key takeaways
– DCB0129 governs the manufacturer of a health IT system’s clinical risk management; DCB0160 governs the deploying organisation (e.g. an NHS trust). Both are mandatory under the UK Health and Social Care Act 2012, and both require a named Clinical Safety Officer (CSO). The current editions (DCB0129 v4.2 / DCB0160 v3.2) are under an NHS England review, with public consultation open 29 June–11 September 2026.
– HIPAA requires a BAA with any vendor that creates, receives, maintains, or transmits PHI on a covered entity’s behalf — including AI/model vendors. But not every AI vendor’s standard BAA covers every modality (audio, fine-tuning data, prompt/log retention). Verify scope per vendor. The BAA is only the contracting half; HIPAA’s Security Rule technical safeguards (45 CFR §164.312) are the half your engineering team builds.
– The EU AI Act classifies certain medical/health AI as high-risk, adding conformity-assessment and governance obligations on top of medical-device law. Obligations phase in over several years: use-based (Annex III) high-risk duties apply from 2 December 2027, and product-embedded/medical-device (Annex I) duties from 2 August 2028.
– In the US, AI/ML-enabled medical devices with a diagnostic or treatment function fall under FDA’s SaMD framework, with Predetermined Change Control Plans (PCCPs) for models that adapt after clearance. This applies to FDA-regulated device functions, not general workflow automation.
– None of these frameworks are satisfied by the AI model alone. They’re satisfied by the system around it — and that system is the engineering deliverable where most deployments actually stall.
– Trembit is not a Clinical Safety Officer and does not train diagnostic models. Trembit builds and integrates the compliant platform and audit/governance architecture that helps a client’s CSO and compliance team meet these standards.New to where clinical AI actually stands today? Start with our companion guide to the broader landscape of what’s production-ready in healthcare AI today — this piece picks up where that one leaves off, at the compliance and deployment layer.
What Is Clinical AI, and Why Does It Need Its Own Compliance Framework?
For the purposes of this guide, clinical AI is any AI system that touches patient data or a clinical or healthcare-operational workflow — from an ambient-documentation assistant and a triage summariser to a risk-scoring model feeding an EHR. That’s a deliberately narrow definition, and it matters, because it draws the line where the compliance bar changes.
Generic business AI carries commercial and data-protection risk. Clinical AI carries those plus two things ordinary software doesn’t: direct patient-safety consequences and, in the UK, a statutory clinical risk management duty that simply doesn’t exist for non-health software. A wrong output isn’t a bad quarter — it can be a wrong record on a patient chart. That’s why the same LLM you’d deploy for internal ops can’t be dropped into a clinical pathway on the same terms.
The rest of this guide uses a three-layer framing, because most real deployments have to answer all three at once:
- Clinical safety — DCB0129 / DCB0160 (UK).
- Data protection — HIPAA (US) and GDPR (EU/UK).
- Emerging AI-specific governance — the EU AI Act and FDA’s SaMD framework.
The frameworks overlap, but none replaces another. A HIPAA BAA does nothing for your DCB0160 obligation; a DCB0129 safety case says nothing about your PHI handling in the US.
What Is DCB0129, and Who Does It Apply To?
DCB0129 is the UK standard for “Clinical Risk Management: its Application in the Manufacture of Health IT Systems.” Published by NHS England Digital, it governs the manufacturer — the organisation that develops the health IT or software system, including AI and software vendors building for the NHS or a UK health IT context. Compliance is mandatory under the Health and Social Care Act 2012, which gives NHS information standards their statutory force.
Under DCB0129, the manufacturer has to do four concrete things, not just write a policy:
- Appoint a named Clinical Safety Officer (CSO) — a suitably qualified, registered clinician responsible for clinical safety on the manufacturer side.
- Establish a Clinical Risk Management System (CRMS) — the documented process governing how clinical risk is identified and managed across the product’s development.
- Maintain a hazard log throughout development — a living register of identified clinical hazards, their causes, mitigations, and residual risk.
- Produce a Clinical Safety Case Report before release — the documented argument, backed by evidence, that the system is safe to deploy.
A note on currency, because it matters on a topic this precise: the current editions are DCB0129 v4.2 and DCB0160 v3.2, both published in 2018 and both currently under a formal NHS England review, with a public consultation running 29 June to 11 September 2026. The commonly used reference names “DCB0129” and “DCB0160” are what NHS procurement and tenders still cite, but confirm the current designation at the time you build — this is exactly the kind of standard that gets reissued.
What Is DCB0160, and How Does It Differ From DCB0129?

DCB0160 is the deployment-side counterpart: “Clinical Risk Management: its Application in the Deployment and Use of Health IT Systems.” It governs the deploying organisation — the NHS trust, primary care provider, or health system that puts the software into live clinical use — not the vendor that built it.
This is the single most common confusion point for teams new to these standards, so state it plainly: a manufacturer’s DCB0129 sign-off does not discharge the deployer’s DCB0160 obligations. The deploying organisation needs its own named CSO and its own clinical safety case, covering how the system is configured, integrated, and used in their specific environment — with their patient population, their workflows, and their existing systems. The same product deployed in two trusts needs two DCB0160 safety cases, because the clinical risk lives partly in how it’s used, not only in how it’s built.
In practice both standards operate together: the manufacturer’s DCB0129 evidence (hazard log, safety case, known residual risks) becomes an input the deployer’s CSO relies on to complete the DCB0160 assessment. Typically both are required for a health IT system to go live in an NHS setting. For an engineering team, the practical consequence is that you have to produce artefacts two different CSOs will read: the ones your product’s safety case needs, and the ones your client’s deployment safety case needs.
Does HIPAA Cover AI Vendors, and Where’s the Gap?
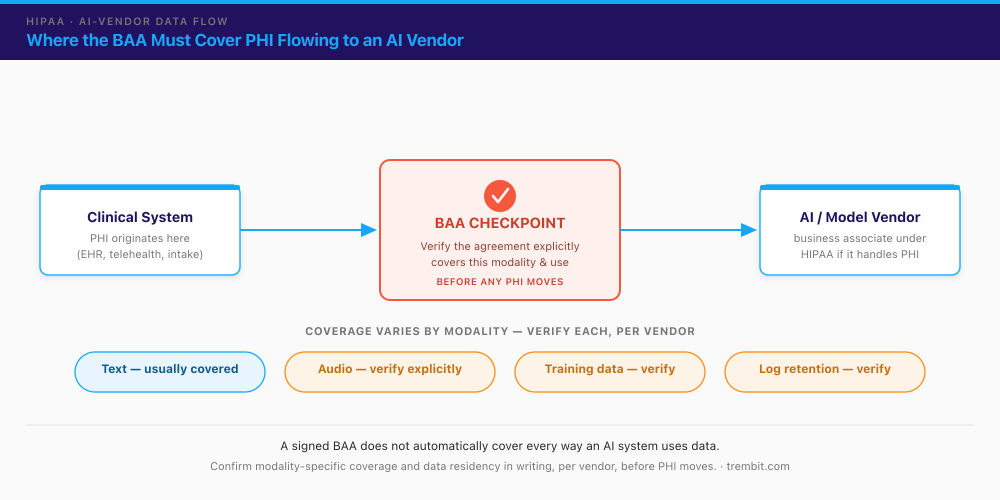
Yes — HIPAA covers AI vendors when they handle PHI. A covered entity (a provider or health plan) must have a signed Business Associate Agreement (BAA) with any vendor that creates, receives, maintains, or transmits protected health information on its behalf. A software or AI vendor processing PHI is a business associate, and the BAA is not optional. Alongside the BAA, two long-standing HIPAA principles apply directly to AI pipelines: minimum necessary (send only the PHI the task actually requires) and de-identification (strip identifiers where the use case allows, especially for training or evaluation data).
The gap — and it’s a real architecture and procurement risk, not FUD — is that a signed BAA does not automatically cover every way an AI system uses data. A general BAA written for text processing may say nothing explicit about audio, about fine-tuning or training a model on customer data, or about how long prompts and model logs are retained. Modern AI systems frequently ingest and retain PHI at scale to train or continually improve models — a use that falls outside HIPAA’s permitted purposes unless it’s explicitly authorised and controlled. HHS published a proposed update to the HIPAA Security Rule in January 2025, with a final rule anticipated but not issued as of mid-2026 — so the current guardrail is the existing BAA framework applied carefully, not an AI-specific regulation.
The practical rule: verify BAA scope per vendor and per data modality before any PHI moves. Confirm in writing that the agreement covers the specific modalities you use, prohibits training on your PHI without explicit authorisation, and defines retention and deletion. The audio modality is where this bites hardest — an ambient clinical-scribe tool sending voice to a model provider is one common place a text-oriented BAA quietly leaves a gap. If you’re solving general telehealth HIPAA compliance for video and EHR infrastructure as well, our guide to HIPAA-compliant AI integration covers that adjacent ground.
The BAA is only the contracting half of HIPAA — the Security Rule is the engineering half. A signed agreement means nothing if the system underneath it doesn’t implement HIPAA’s Security Rule technical safeguards (45 CFR §164.312): access control (unique user IDs, automatic logoff, encryption of PHI at rest), audit controls (mechanisms that record and examine activity in systems handling PHI), integrity controls (protection against improper alteration or destruction of PHI), and transmission security (encryption of PHI in transit). These are the parts your engineering team owns, not legal or procurement. Note that HHS’s proposed 2025 Security Rule update would move several of these from “addressable” to explicitly required — encryption and multi-factor authentication among them — so anyone scoping this work now should build to the stricter bar rather than the current minimum.
Data residency is the AI-vendor-selection criterion most teams discover too late. Where a model’s inference endpoint physically runs is a live procurement blocker, not a footnote: if you’re NHS- or EU-facing and your model vendor serves inference from US-hosted infrastructure, that’s a direct DCB0160, GDPR, and data-residency conflict that surfaces the moment procurement or an information-governance lead reviews the data-flow diagram. Confirm the region a vendor processes and stores PHI in — and whether a region-locked or in-country deployment option exists — before you commit to it, not after the integration is built.
One cross-reference for EU/UK health data: GDPR reaches the same destination by a different legal route — data minimisation, purpose limitation, and a lawful basis for processing special-category health data, formalised in a Data Processing Agreement (DPA) under GDPR Article 28 (the EU counterpart to the BAA). The mechanics differ from HIPAA, but the spirit (only the data you need, only for the purpose you stated) is the same. A full GDPR comparison is out of scope here; see our telehealth security and compliance overview for the cross-jurisdiction picture.
If you’re weighing an AI vendor against these criteria right now and want a second set of eyes on the architecture, talk to our engineering team — it’s the kind of review we do before a line of integration code gets written.
What Other Regulations Govern Clinical AI in 2026? (EU AI Act, FDA, NICE/MHRA)
This section is a landscape pointer, not a deep dive — DCB0129/0160 and HIPAA above are where the depth lives. The AI-specific layer is newer, moving fast, and worth tracking rather than memorising.
EU AI Act (Regulation (EU) 2024/1689). The Act classifies certain AI systems as high-risk, and AI that functions as, or as part of, a regulated medical device generally lands in that category — when a device is class IIa/IIb/III under the MDR (or B–D under the IVDR) and relies on AI, its device classification effectively pulls it into the AI Act’s high-risk bracket. High-risk status triggers conformity-assessment, risk-management, and documentation obligations (Article 43), which can often be run alongside an existing MDR assessment. Timing matters here: the Act phases obligations in over several years. Under the EU’s May 2026 Digital Omnibus agreement, use-based (Annex III) high-risk obligations apply from 2 December 2027, and product-embedded/medical-device (Annex I) high-risk obligations from 2 August 2028. Do not state a blanket “the EU AI Act is in effect for medical AI” without checking which provisions apply on the date you publish.
FDA (US). AI/ML-enabled software with a diagnostic or treatment function is regulated as Software as a Medical Device (SaMD). For models that change after clearance, the FDA’s mechanism is the Predetermined Change Control Plan (PCCP) — a pre-authorised description of what the model may change, how those changes are validated, and how they’re monitored, so certain updates don’t require a new marketing submission. The sequence matters if you’re citing it: the FDA, Health Canada, and the UK’s MHRA jointly published guiding principles for PCCPs on 24 October 2023, and the FDA then finalised its own PCCP marketing-submission guidance in December 2024 — the guidance came after the trilateral principles, not before. (Both build on the earlier Good Machine Learning Practice principles the same three regulators issued in October 2021 — a related but separate document; don’t conflate them.) Crucially, all of this applies to FDA-regulated device functions — not to general clinical-workflow or administrative AI, which is a common and important scoping distinction.
NICE / MHRA (UK). The MHRA’s Software and AI as a Medical Device programme is the UK’s parallel to FDA’s SaMD work, and NICE’s evidence-standards frameworks inform how AI-enabled health technologies are evaluated for use in the NHS. For a clinical-AI build, these mostly matter when the system has a device-like clinical function rather than a workflow-support one.
Emerging, non-statutory. The Coalition for Health AI (CHAI) is an industry-led assurance initiative gaining traction in the US. It is not a regulator and not a statutory requirement — but its assurance-lab and model-card work is worth watching as a possible de-facto expectation from health-system buyers.
What Does It Take to Deploy Clinical AI Safely? (The Deployment Safety Checklist)
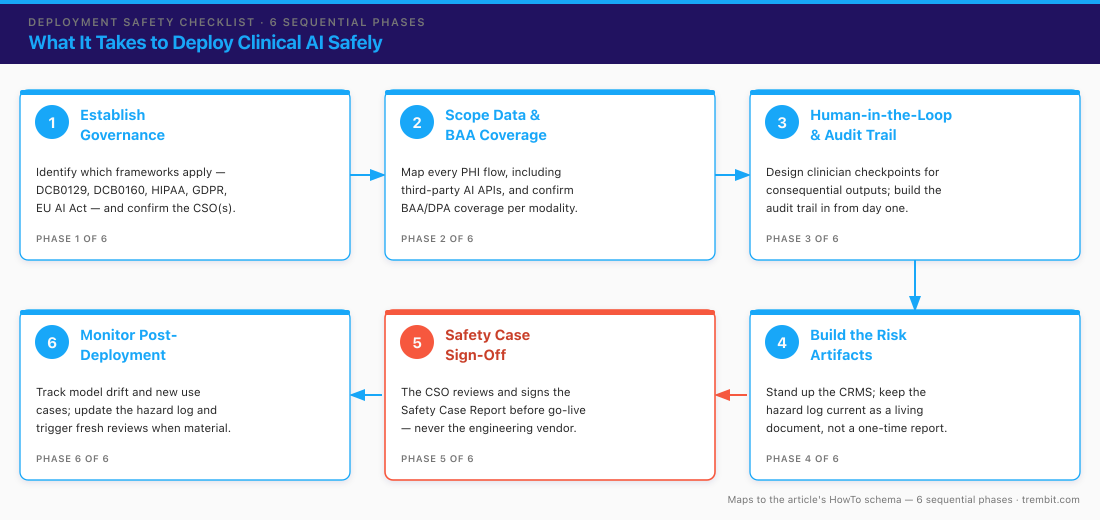
This is a working checklist, not a marketing list. The phases are sequential on purpose — each one produces something the next depends on — and the items are meant to be literally checkable. Hand it to your engineering and product leads.
Phase 1 — Establish governance before you write code
Compliance designed in is cheap; compliance retrofitted is where timelines and budgets blow up. Settle scope and ownership before the first sprint.
- [ ] Determine which frameworks apply to this specific system and jurisdiction: DCB0129 (you’re the manufacturer), DCB0160 (you’re the deployer), HIPAA (US PHI), GDPR (EU/UK health data), EU AI Act (regulated medical function in the EU), FDA SaMD (diagnostic/treatment function in the US).
- [ ] Identify and confirm the named Clinical Safety Officer(s) involved — yours if you’re a manufacturer or deployer, or your client’s. (Trembit does not fill this role.)
Don’t have a Clinical Safety Officer yet? That’s common at Series A/B, and it isn’t a reason to stall the engineering. DCB0129/0160 require a named, suitably qualified CSO — usually a registered clinician, engaged in-house or on a fractional/contract basis — and that appointment is the client’s to make, not ours. What we can do is build the system so that when your CSO arrives (or is retained), the evidence they need already exists: the hazard-log inputs, audit trails, and safety-case artefacts are in place rather than reconstructed after the fact. We work alongside your CSO; we never stand in for one.
– [ ] Decide, in writing, whether the system has a device-like clinical function (which pulls in FDA/EU-AI-Act device obligations) or is workflow/operational support (which usually doesn’t). This one decision reshapes the whole plan.
Phase 2 — Scope the data and confirm BAA / data-protection coverage
You can’t protect data whose path you haven’t mapped. Do this before any PHI moves, not after the integration is built.
- [ ] Map every place PHI or clinical data flows, including into and out of any third-party AI or model API.
- [ ] Confirm a BAA (or GDPR-equivalent) explicitly covers every modality in use — text, audio, images, fine-tuning/training data, and prompt/log retention — not just “the vendor is HIPAA-eligible.”
- [ ] Define de-identification and minimum-necessary rules per data flow before data moves; confirm the vendor is contractually barred from training on your PHI without explicit authorisation.
Phase 3 — Architect for human-in-the-loop and auditability from day one
Auditability bolted on after the fact is expensive and usually incomplete. Build it into the data model.
- [ ] Design the checkpoint where a qualified clinician or reviewer approves consequential AI outputs before they act on a patient record — no autonomous write-through on clinically significant actions.
- [ ] Build the audit trail into the system, not as an afterthought: capture what the AI saw (inputs), what it produced (outputs and, where possible, rationale/confidence), the model/version, and who approved or overrode it.
- [ ] Make outputs traceable and explainable enough to review — a reviewer must be able to see why a recommendation appeared, not just the recommendation.
Phase 4 — Build and maintain the risk artifacts
In a DCB0129/0160 context these are living documents, not a one-time report filed at launch.
- [ ] Stand up the Clinical Risk Management System (CRMS) as the governing process for the whole development lifecycle.
- [ ] Keep a hazard log current: every identified clinical hazard, its cause, mitigation, and residual risk after mitigation.
- [ ] Update the artifacts as the system changes — new features, new data sources, and new failure modes all generate new hazards.
Phase 5 — Produce the Clinical Safety Case Report and get sign-off
The safety case is the evidence-backed argument that the system is safe to deploy. It is reviewed and signed by a CSO — never by the engineering vendor.
- [ ] Assemble the Clinical Safety Case Report for review by the manufacturer’s and/or deployer’s CSO before go-live.
- [ ] Supply the CSO the evidence they need: test results, audit-log samples, architecture documentation, the hazard log, and residual-risk statements.
- [ ] Confirm the sign-off authority stays with the named CSO. Trembit’s role is producing the evidence, not signing the safety case.
Phase 6 — Monitor, re-assess, and keep the hazard log current post-deployment
Clinical risk management does not end at go-live. This is where “learning” AI models create obligations ordinary software doesn’t.
- [ ] Monitor for model drift, new use cases, and configuration changes, each of which can introduce new clinical risk.
- [ ] Update the hazard log whenever the operating context changes, and trigger a fresh safety-case review where the change is material.
- [ ] For adaptive AI/ML under FDA, keep changes within the authorised PCCP envelope; changes to intended use fall outside it and need fresh regulatory review.
Who Is Responsible for What — the Manufacturer, the Deploying Organisation, and Your Engineering Team?
Three distinct roles run through everything above, and keeping them separate is what keeps a project honest.
The manufacturer carries the DCB0129 obligations. It has a named CSO, runs the CRMS through development, maintains the product hazard log, and owns the Clinical Safety Case Report for the product itself.
The deploying organisation carries the DCB0160 obligations. It has its own named CSO and its own safety case covering how the product is configured and used in its environment. It relies on the manufacturer’s evidence but signs its own case.
The engineering partner — Trembit’s role — builds the compliant architecture, audit trails, data pipelines, and human-in-the-loop controls that give both CSOs the evidence they need. To be unambiguous: Trembit does not sign a Clinical Safety Case Report, does not hold FDA clearance or a CE mark, and does not train the diagnostic model. Trembit is the team that makes the system those CSOs are reviewing actually meet the standard. Where a workflow involves clinical review steps or automated agents acting under human supervision, that’s built with the same auditable-by-design pattern the checklist above describes — AI agents constrained by human-in-the-loop controls, running under governed orchestration.
How Does Trembit Help Teams Meet These Frameworks?
Trembit’s relevant credential isn’t a diagnostic model — it’s a track record of building healthcare and high-stakes software to audit-grade discipline: encryption, access control, traceability, and uptime that holds up under review.
One honest caveat before the proof points, because a sharp reader will notice it otherwise: we haven’t yet publicly named a clinical-AI-specific engagement that took an AI system through a DCB0129/0160 safety case. What we can show is the two halves that combine into exactly that capability — audit-grade healthcare compliance engineering (Martti, WebPRAX) and explainable, human-in-the-loop AI in a regulated domain (Eska) — under real certification and compliance regimes.
Equiti Health / Martti. Trembit engineered a WebRTC medical video-interpretation platform used across 4,000+ healthcare facilities, with integrations into Epic, Oracle Cerner, and eClinicalWorks EHRs, HIPAA and SOC 2 compliance, and 99.99% uptime. This is evidence of the exact engineering discipline clinical-AI compliance architecture demands — encryption, access control, auditability, EHR integration under a real compliance regime — not an AI or diagnostic claim. See the Martti medical video-interpretation case study.
WebPRAX (Germany). Trembit built a KBV-certified video platform for psychotherapy, with Germany-only data residency and DSGVO (GDPR) compliance. KBV is a different specific standard, but taking a healthcare product through a national regulator’s certification process is the same category of rigour that DCB0129/0160 and HIPAA demand — see the P2P psychotherapy platform case study, and, for the certification detail, our note on meeting KBV certification requirements.
Eska (FinTech leasing — cross-industry pattern). Outside healthcare, Trembit built an explainable ML risk-scoring pipeline with a human-review workflow, full decision traceability, and ERP integration. It’s a direct proof of the engineering pattern this piece’s checklist calls for — auditable, explainable, human-in-the-loop, logged — applied in a regulated domain. See the Eska leasing AI risk platform case study. (Eska is a FinTech project, not a healthcare one — cited only as proof of the pattern.)
If you’re scoping a clinical AI deployment and need the architecture to pass a DCB0129/0160 or HIPAA review, Trembit’s team can work alongside your Clinical Safety Officer to build it right — book a call with our engineering team and bring the data-flow diagram.
FAQ
What is DCB0129?
DCB0129 is the UK standard “Clinical Risk Management: its Application in the Manufacture of Health IT Systems.” It applies to the manufacturer — the organisation that develops a health IT or software system, including AI vendors building for the NHS. It’s mandatory under the Health and Social Care Act 2012 and requires the manufacturer to appoint a named Clinical Safety Officer, run a Clinical Risk Management System, maintain a hazard log throughout development, and produce a Clinical Safety Case Report before release. The current edition is v4.2 (2018), under NHS England review through 2026.
What is DCB0160, and how is it different from DCB0129?
DCB0160 is the deployment-side counterpart — “Clinical Risk Management: its Application in the Deployment and Use of Health IT Systems.” Where DCB0129 governs the vendor that built the system, DCB0160 governs the organisation deploying it into clinical use (e.g. an NHS trust). The deployer needs its own CSO and its own safety case covering how the system is configured and used in its environment; a manufacturer’s DCB0129 sign-off doesn’t discharge it. Both are typically required for a system to go live in NHS/UK health IT.
Does HIPAA cover AI vendors?
Yes — if an AI vendor creates, receives, maintains, or transmits PHI on a covered entity’s behalf, a signed Business Associate Agreement (BAA) is required. The important caveat is that coverage varies by vendor and by data modality: a general BAA may not explicitly cover audio, fine-tuning on customer data, or prompt/log retention. Don’t assume a standard BAA covers every use of an AI system — verify scope per vendor and per modality, in writing, before sending PHI. And remember the BAA is only the contracting half: the system still has to implement HIPAA’s Security Rule technical safeguards (45 CFR §164.312 — access control, audit controls, integrity, transmission security), which are the engineering team’s responsibility.
Is Trembit a Clinical Safety Officer, or does Trembit provide regulatory sign-off?
No. Trembit is the engineering partner that builds the compliant platform and the evidence — audit trails, architecture documentation, test results, hazard-log inputs — that a client’s own CSO uses to complete their DCB0129/0160 safety case. The sign-off authority stays with the client’s named Clinical Safety Officer. Trembit does not sign the Clinical Safety Case Report, does not hold FDA clearance or a CE mark, and does not train diagnostic models.
How is the EU AI Act different from DCB0129/0160 or HIPAA?
They operate on different axes. DCB0129/0160 are UK clinical-risk-management standards for health IT specifically. HIPAA is US data-protection law for PHI. The EU AI Act is a horizontal AI regulation that classifies certain medical AI as high-risk and layers AI-specific obligations — risk management, technical documentation, conformity assessment — on top of, not instead of, existing medical-device and data-protection law. A system can be subject to all three at once. The AI Act’s high-risk obligations phase in over time: 2 December 2027 for use-based (Annex III) systems and 2 August 2028 for product-embedded/medical-device (Annex I) systems.
How long does it take to get a clinical AI system through DCB0129/0160 or HIPAA compliance?
It depends heavily on scope and, more than anything, on whether compliance was designed in from the start or retrofitted. Retrofitting audit trails, human-in-the-loop checkpoints, and data-governance controls into a system that wasn’t built for them is where timelines and budgets blow up — the same integration bottleneck that stalls most healthcare-AI deployments. We won’t put a fabricated number on it; the honest answer is that designing the governance and audit architecture into Phase 1 is what keeps the schedule predictable — which is why teams that loop us in before the architecture is locked get a scope-based estimate instead of a guess. Talk to us early.
Deploying clinical AI safely is a compliance-and-engineering problem, not a modeling problem. DCB0129/0160, HIPAA, and the emerging AI-specific layer all point to the same underlying requirement: a documented, auditable, human-in-the-loop system architecture. That’s the deliverable — and it’s the work Trembit does.
If you’re scoping a clinical AI deployment and want the architecture built to pass a DCB0129/0160 or HIPAA review the first time, talk to our engineering team. We’ll work alongside your Clinical Safety Officer, map the data flows and BAA/DPA coverage, and design the audit and human-in-the-loop controls in from Phase 1 — before a retrofit becomes the expensive option.